CAPR - Capricor Therapeutics Sarepta Therapeutics: A Promising Clinical-Stage Biotech And A Commercial-Stage Leader In Duchenne Muscular Dystrophy
2023-10-25 12:39:22 ET
Summary
- Recent dip in CAPR's stock price following update on regulatory pathway for CAP-1002 and cash raise represents a buying opportunity.
- Completion of enrollment of Cohort A of ongoing Phase 3 trial of CAP-1002 is expected in Q4 2023, topline results in Q4 2024 and BLA submission in 2025.
- Risks for CAPR include; failure of ongoing Phase 3 trial, regulatory delays and need for more dilutive cash raise by the end of 2024.
- SRPT is a commercial-stage leader in the DMD field, with four approved genetic therapies, great cash balance, increasing revenues and promising pipeline.
- Long-term risks for SRPT include failure of ongoing confirmatory trials and competitions by several biotechs developing genetic therapies for DMD.
Thesis overview
Capricor Therapeutics' ( CAPR ) lead asset is CAP-1002, a cell therapy for Duchenne Muscular Dystrophy ("DMD"), currently in the Phase 3 stage of its clinical development. An interim analysis is anticipated in Q4 2023, topline results in Q4 2024, and BLA submission in 2025. Considering promising results in two Phase I/II studies (one published in Lancet ) I believe there is a high probability that the ongoing Phase 3 study will be successful and sufficient (considering dismal prognosis for DMD patients with current therapies) for approval. Despite availability of several treatment options (including glucocorticoids and five approved genetic therapies in the US) there is still significant room for new/better treatments and CAPR believes (and I agree) that CAP-1002 has the potential to be used as a backbone therapy (i.e., in combination with other available treatments). CAPR's stock price has recently dipped by >50% following the announcement of the planned regulatory pathway (estimated BLA submission in 2025 vs. investors' expectation for a much earlier accelerated approval) and a $23M offering . This represents a good buying opportunity in my opinion.
Beyond CAP-1002, CAPR is also developing a proprietary exosome platform. CAPR is exploring its deployment in two broad modalities: precision therapeutics (targeted delivery of therapeutic molecules by exosomes) and vaccinology. This platform, albeit interesting, is still in the preclinical stage and will not be discussed in this article. To advance the exosome platform CAPR is currently focused on securing partners.
Although the main focus of the article is CAPR, much of the discussion below is also very relevant to Sarepta Therapeutics' ( SRPT ) DMD pipeline. SRPT is a leading developer of DMD therapies, including four FDA-approved genetic therapies and more in the pipeline. Furthermore, SRPT is also developing gene therapies for Limb-girdle muscular dystrophy ("LGMD"), which however will not be the focus of this article. In contrast to CAPR, SRPT is a commercial-stage biotech with a good cash balance and increasing revenues. Considering complementary mechanism of action CAP-1002 would likely be used as an add-on to SRPT's genetic therapies.
CAPR's pipeline (CAPR website)
{kind=link}
SRPT's pipeline (SRPT website)
{kind=link}
Overview of DMD
Duchenne muscular dystrophy is caused by a defective gene located on the X chromosome that is responsible for the production of dystrophin. Dystrophin provides mechanical reinforcement in muscle fibers. Its absence results in degeneration of muscle fibers and muscle weakness. Much of the damage in DMD is attributable to a maladaptive immune response (hence the role of glucocorticoids and CAP-1002). Duchenne muscular dystrophy is the most severe of the dystrophinopathies, while other pathogenic variants of the dystrophin gene are associated with later onset and/or milder presentations (Becker muscular dystrophy and intermediate phenotypes).
Being an X-linked genetic disorder DMD typically affects males. Clinically apparent symptoms occur in approximately 22% of female carriers of a mutated dystrophin gene, but are typically milder. Very rarely (about 1 in 50 million live female births) a full-blown presentation similar to males occurs in females (typically associated with mutations in both genes).
With DMD , the clinical onset of weakness usually occurs at the age of 2-3 years. Additional features include cardiomyopathy, respiratory failure, bone fractures, and scoliosis. Affected children frequently have varying degrees of mild cognitive impairment. Patients may experience some improvement between 3-6 years of age. However, this is followed by gradual deterioration. Patients often require a wheelchair by about age 12 to 13 years and die in their late teens or twenties from respiratory insufficiency or cardiomyopathy. Rarely, patients survive in their early thirties. Disease modifying treatments (to be discussed below) can delay disease progression resulting in a median life expectancy of about 30 years . Survival beyond the 4th decade remains uncommon.
Expected clinical course of DMD (CAPR presentation)
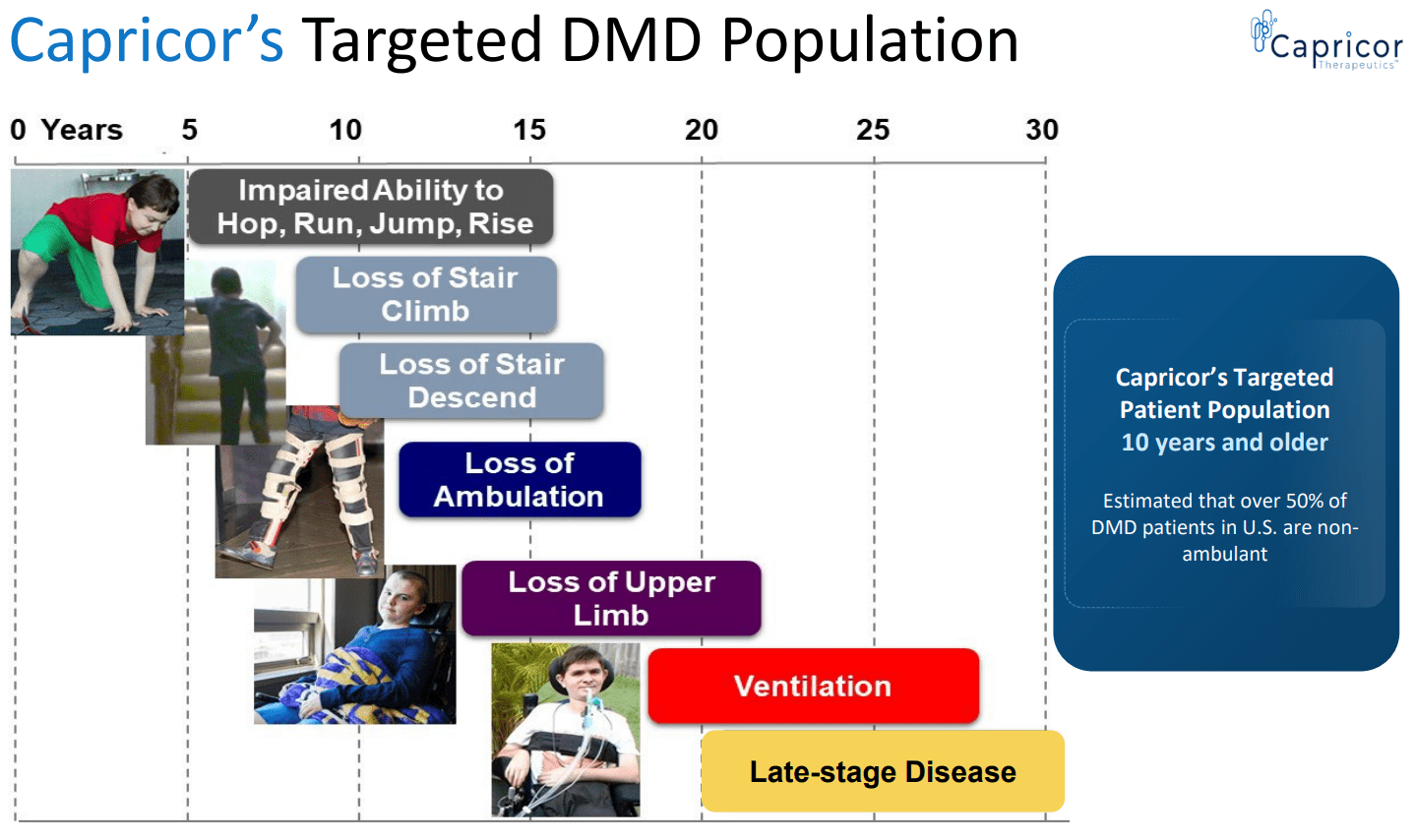{kind=link}
Overview of genetics in DMD
Note; There is some medical jargon below. This is useful to understand the mechanism of action of available treatment options but if you have limited time I would skip to the next section.
There are various pathogenic variants of the DMD gene. Most common (about 70%) mutations are deletions of one or more exons ( exon = part of a gene that will form a part of the final mature RNA produced by that gene after introns have been removed by RNA splicing). Partial gene duplications occur in about 10% of patients. The DMD pathogenic variants in the remaining patients (i.e., without detectable deletions or duplications) are single nucleotide variants, small deletions or insertions, or splice site variants.
Simplified illustration of the type of mutations in the DMD gene (Duchenne.com)
Simplified illustration of RNA splicing. Genes contain exons (which code parts of the final mRNA product) and introns (which are removed by a process called splicing) ("What is RNA splicing?" from Yourgenome.org)
The phenotype of dystrophinopathies is primarily dependent upon the quantity and quality of residual dystrophin in muscle, with absent or minimal dystrophin (e.g., 0-5% of normal) correlating with a more severe phenotype ("DMD") and higher levels of dystrophin (5-50% of normal) correlating with less severe phenotypes (intermediate or Becker muscular dystrophy). DMD is typically associated with out-of-frame deletions, i.e., mutations that shift the translational reading frame resulting in unstable messenger RNA ("mRNA") and the production of a severely truncated dystrophin molecule, which is rapidly degraded in the cell.
Simplified illustration of the implication of out-of-frame (severe disease) vs in-frame (mild disease) mutations (Brenner Computational Biology Research Group)
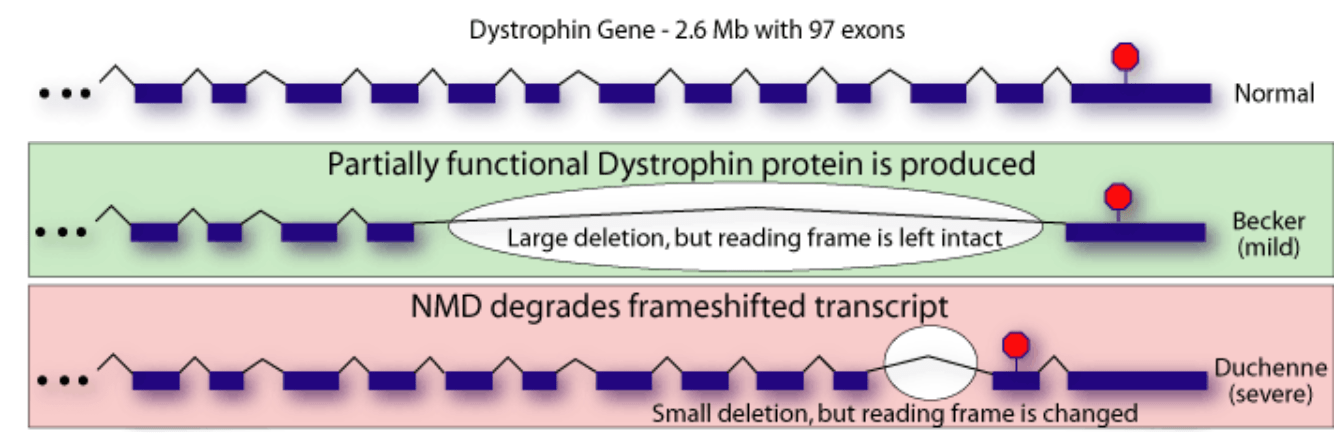{kind=link}
Overview of current treatment options
Disease-modifying treatment includes glucocorticoids and genetic therapies. Glucocorticoids can improve muscle strength, delay disease progression, delay the onset of cardiomyopathy and prolong survival. However, benefit is moderate, delaying age at loss of ambulation by 1.4-2.5 years (maybe longer ). Even with glucocorticoid treatment more than half lose ability to ambulate by about 15 years old and majority by about 20 years old. Furthermore, long-term glucocorticoid therapy is associated with major side effects , including weight gain, decreased linear growth and short stature, hirsutism, and cushingoid appearance (as well as numerous other side effects).
Currently available genetic therapies have the following mechanisms of action;
- Exon skipping with antisense oligonucleotides (eteplirsen, golodirsen, viltolarsen). See here for an illustration of how exon skipping works. Briefly, these therapies aim to restore the reading frame and therefore production of shorter but functional dystrophin. Although this approach does not restore the normal dystrophin protein, it is a reasonable approach considering the milder clinical presentation in patients with a partially functional dystrophin (as seen in patients with Becker and intermediate phenotypes).
- Read-through of premature termination codon (ataluren); Allows increased expression of full-length dystrophin in DMD patients with nonsense mutations.
- Microdystrophin transgenes delivered by adeno-associated virus vectors (delandistrogene moxeparvovec); It is designed to deliver the microdystrophin transgene to skeletal and cardiac muscles.
Importantly, as explained by UpToDate authors, these "therapies increase dystrophin expression, but clinical benefit has not been established". In other words approval of above genetic therapies was based on their ability to increase dystrophin (which is reasonable considering the pathophysiology of the disease discussed above, and milder presentation in patients with Becker and intermediate phenotypes which are associated with at least some functional dystrophin). A more detailed discussion of these therapies follows in the next section.
Overview of genetic therapies
If you have limited time you may skip to the next section.
The following genetic therapies have been approved;
- Eteplirsen (by Sarepta Therapeutics); An antisense oligonucleotide approved for patients who have a confirmed deletion of the DMD gene amenable to exon 51 skipping (13% of DMD patients). Administered intravenously ("IV") once weekly. Notably, eteplirsen was approved in 2016 based on a 24-week placebo-controlled trial with just n=12 patients (ages 7 to 13 years and ambulatory), based on the accelerated approval pathway and the surrogate outcome of increased dystrophin production. Of interest is that " although the FDA initially set a November 2020 deadline for eteplirsen's manufacturer to complete a clinical trial determining whether the drug has clinical benefit, the company will not complete the trial until 2024 or later".
- Golodirsen (by Sarepta Therapeutics); An antisense oligonucleotide approved for patients who have a confirmed deletion of the DMD gene amenable to exon 53 skipping (8% of DMD patients). Administered IV once weekly. Approved by the FDA in 2019 based upon the surrogate outcome of increased dystrophin production. Approval was based on a small (n=12 patients) placebo-controlled study and an open-label extension (including n=13 additional patients). "After 48 weeks or more of treatment, the mean dystrophin level increased from 0.1 percent of normal at baseline to 1.02 percent of normal. Clinical benefit was not reported". Proof of benefit in clinical outcomes (ongoing study) is required for full-approval.
- Viltolarsen (by Nippon Shinyaku); An antisense oligonucleotide approved for patients who have a confirmed deletion of the DMD gene amenable to exon 53 skipping (8% of DMD patients). Administered IV once weekly. Approved by the FDA in 2020 based upon the surrogate outcome of increased dystrophin production. Approval was based on a small (n=16) dose-finding safety trial. "Patients assigned to viltolarsen 80 mg/kg once a week showed an increase in dystrophin from a mean level of 0.6 percent of normal at baseline to 5.9 percent of normal at week 25." Similar to above exon-skipping therapies, continued FDA approval depends upon confirmation of clinical benefit in ongoing trials.
- Casimersen (by Sarepta Therapeutics); An antisense oligonucleotide approved for patients who have a confirmed deletion of the DMD gene amenable to exon 45 skipping (8% of DMD patients). Administered IV once weekly. Approved by the FDA in 2021 based upon the surrogate outcome of increased dystrophin production. Approval was based on a 48-week placebo-controlled randomized trial with n=43 patients. "At 48 weeks, the mean increase in dystrophin levels from baseline on muscle biopsy for patients assigned to casimersen was 0.81 percent, compared with 0.22 percent for the placebo group"
- Ataluren (by PTC Therapeutics); An orally administered drug conditionally approved in Europe for the approximately 10-15% of DMD patients with nonsense (stop) mutations (i.e., mutations that cause a protein to end its translation earlier than expected, resulting in a truncated non-functioning protein). Ataluren promotes ribosomal read-through of nonsense mutations, allowing bypass of the nonsense mutation and continuation of the translation process to production of a functioning protein. Ataluren has been rejected by the FDA and faces potential withdrawal from Europe as well. Despite increased full-length dystrophin expression in early small trials and signals of clinical benefit, larger trials have failed to prove benefit in clinical outcomes.
- Delandistrogene moxeparvovec (by Sarepta Therapeutics); An adeno-associated virus capsid containing a transgene encoding a microdystrophin protein under the control of a muscle-specific MHCK7 promoter. It is designed to deliver the microdystrophin transgene to skeletal and cardiac muscles. The rationale for using microdystrophin (vs. full-length dystrophin) is that the gene encoding the full-length dystrophin protein exceeds the packaging capacity of a single AAV vector. Microdystrophin is shortened but functional. Similar to above genetic therapies, delandistrogene moxeparvovec was granted accelerated approval in July 2023, pending confirmatory trials. Currently , it is approved only for ambulatory pediatric patients aged 4 through 5 years, based on a subgroup analysis showing clinical benefit in these patients (lack of statistical significance in children 6-7 years old was attributed to higher baseline severity in the treatment arm vs. placebo). The US prescribing information carries warnings and precautions regarding acute serious liver injury, immune-mediated myositis, myocarditis and pre-existing immunity against AAV serotype rh74 (which may impede transgene transduction at desired therapeutic levels). Delandistrogene moxeparvovec is contraindicated in patients with any deletion in exon 8 and/or exon 9 in DMD (due to the increased risk of a severe immune-mediated myositis reaction) and its use is not recommended in patients with an anti-AAVrh74 total binding antibody titre of ? 1:400 (about 14% would be non-eligible based on just the latter criterion). It is administered intravenously once. The current wholesale acquisition cost is $3.2 million.
Important notes about available genetic therapies
The following are notable for genetic therapies;
- Most are commercialized by Sarepta Therapeutics (eteplirsen, golodirsen, casimersen, delandistrogene moxeparvovec), the only exception being viltolarsen, which is commercialized by Nippon Shinyaku, and ataluren by PTC Therapeutics (which has been rejected more than once by the FDA and faces potential withdrawal from Europe). Of interest is that Nippon Shinyaku has partnered with CAPR as will be discussed below.
- Exon skipping therapies work only for a fraction of DMD patients with amenable mutations; eteplirsen (exon 51 skipping, 13%), golodirsen/viltolarsen (exon 53 skipping, 8%), casimersen (exon 45 skipping, 8%). Ataluren could benefit the estimated 10-15% of DMD patients with nonsense mutations (but is not approved in US and faces potential withdrawal from Europe). Collectively, available exon-skipping therapies and ataluren are suitable for just 39-43% of DMD patients.
- The big deal for SRPT about delandistrogene moxeparvovec is potential for a much broader label. In contrast to exon skipping (suitable only for the small fraction of patients with relevant mutations), delandistrogene moxeparvovec should in theory be useful irrespectively of underlying mutation. However, current label is limited to patients aged 4 through 5 years.
- Available genetic therapies do NOT cure DMD. Pathophysiologically, exon skipping achieves moderate increases in dystrophin (levels much lower than normal). Notably, the produced truncated dystrophin is only partially functional. Delandistrogene moxeparvovec delivers a microdystrophin gene (which encodes a shorter but functional version of dystrophin). Notably, the resulting microdystrophin is smaller than truncated dystrophins produced with exon-skipping therapies. Finally, ataluren increases full-length dystrophin expression, but not to a normal level (plus has failed to produce clinical benefit).
- Approval of genetic therapies was based on the surrogate endpoint of increased expression of dystrophin (which I believe is reasonable as discussed in above sections, and I expect most ongoing trials to confirm clinical benefit), which contrasts approval pathway for CAP-1002 (to be discussed in a later section). Notably, studies that lead to approvals were very small (n=12-43 patients).
- Failure of ataluren to improve clinical outcomes despite increased production of full-length dystrophin expression is concerning for approved exon-skipping therapies.
- Antisense oligonucleotides are administered intravenously once weekly. Obviously, and considering dismal prognosis of DMD, patients don't mind this. Delandistrogene moxeparvovec is given as a single intravenous infusion.
CAP-1002
CAP-1002 is comprised of cardiosphere-derived cells ("CDCs"), which is an endogenous population of stromal cells derived from cells of healthy human hearts. The ability of CAP-1002 to slow disease progression in DMD lies in the immunomodulatory, anti-inflammatory, and anti-fibrotic actions of CDCs, which are mediated by secreted exosomes containing bioactive cargo. CAP-1002 is administered intravenously once every three months. CAP-1002 is manufactured from donor hearts via a proprietary process and has a clinical self-life of over four years. CAP-1002 functions by reducing fibrosis and inflammation via the secretion of exosomes, helping to halt the progression of DMD.
Notably, FDA has granted CAP-1002 regenerative medicine advanced therapy ("RMAT"), orphan drug, and rare pediatric disease designations. Therefore, CAP-1002 is eligible for rolling review and priority review (which also means a priority review voucher upon approval, typically worth around $100 million).
CAP-1002 has completed preclinical studies, 2 randomized ph1/2 studies (HOPE and HOPE 2) and a 24-month open-label extension of HOPE 2. All clinical studies have shown consistent benefits in both cardiac and skeletal muscle function. Treatment was also well tolerated. CAP-1002 is currently being evaluated in a phase 3 (HOPE 3) trial but the regulatory pathway to approval (to be discussed below) is complicated by CAPR's plan to shift to another manufacturing facility for commercialization (vs. the facility that has been used so far for HOPE, HOPE 2 and first cohort of HOPE 3).
HOPE-2 trial
HOPE-2 was a randomized, placebo-controlled Phase 2 study, published in Lancet . Patients were randomized to CAP-1002 or placebo for 12 months. All patients were on corticosteroids and 80% were non-ambulant. The primary outcome was the mid-level Performance of Upper Limb test (PUL version 1.2).
Based on PUL 1.2 the study showed 71% slowing of loss of upper limb function compared to placebo; the change in the primary endpoint at 12 months for the CAP-1002 group was -0.8 points and for the placebo group was -3.4 points, corresponding to a treatment difference of 2.6 points. This was not only statistically significant but also very meaningful for these patients. As explained in the Lancet publication; "A 1-point change in mid-level PUL 1.2 was felt to be clinically meaningful for the original statistical powering of the study because it translates to loss of one critical activity requiring elbow muscle function. For example, it might represent loss of the ability to bring the hands to the mouth or table, to stack, lift, or move objects on a table, or to remove the lid from a container. In non-ambulant patients dependent on upper limb function, such seemingly small declines signal major deterioration. The ability of CAP-1002 to prevent or delay such deterioration would be expected to translate to prolonged independence of patients with Duchenne muscular dystrophy".
Additionally the study showed benefit in several cardiac function markers, "showing a 107% slowing of progression of cardiac disease (p=0.002)". The CAP-1002 group had a mean increase in left ventricle ejection fraction by 0.1% vs. a mean decrease by 3.9% in the placebo group (4% absolute difference). Consistent benefit (albeit non-significant) was also shown in multiple exploratory cardiac MRI structure endpoints (which are surrogate measures of cardiac function and are considered significant in terms of relevance to long-term outcomes). Furthermore, a statistically significant decrease in the proportion of CK-MB (an enzyme specific to cardiac muscle) was shown. The results of the placebo patients were consistent with natural history (worsening), while in the treated group, most patients were stable or improved.
CAP-1002 was generally well tolerated, with the exception of hypersensitivity reactions. Implementation of a pretreatment procedure mid-study succeeded in preventing further serious allergic reactions.
Statistically significant improvement in both upper limb and cardiac results (CAPR presentation)
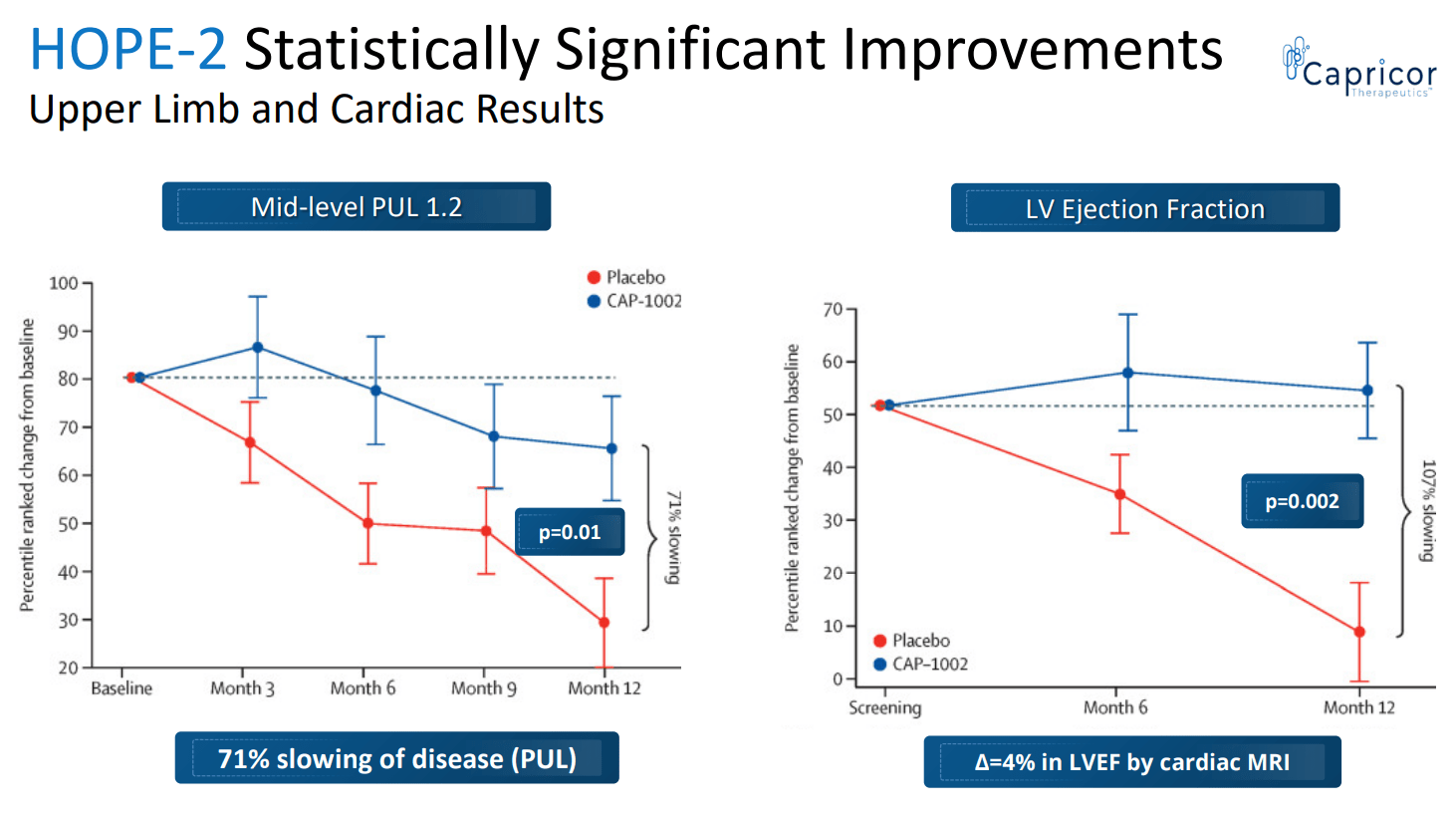{kind=link}
Summary of 12-month results from randomized HOPE-2 trial (SRPT's 10K)
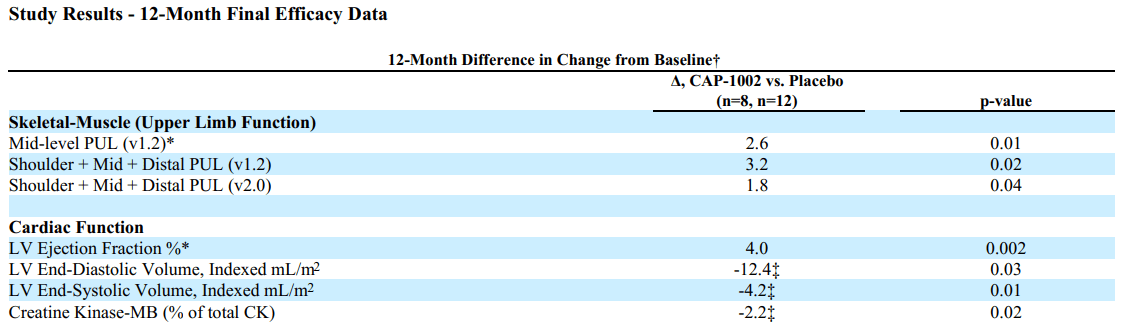{kind=link}
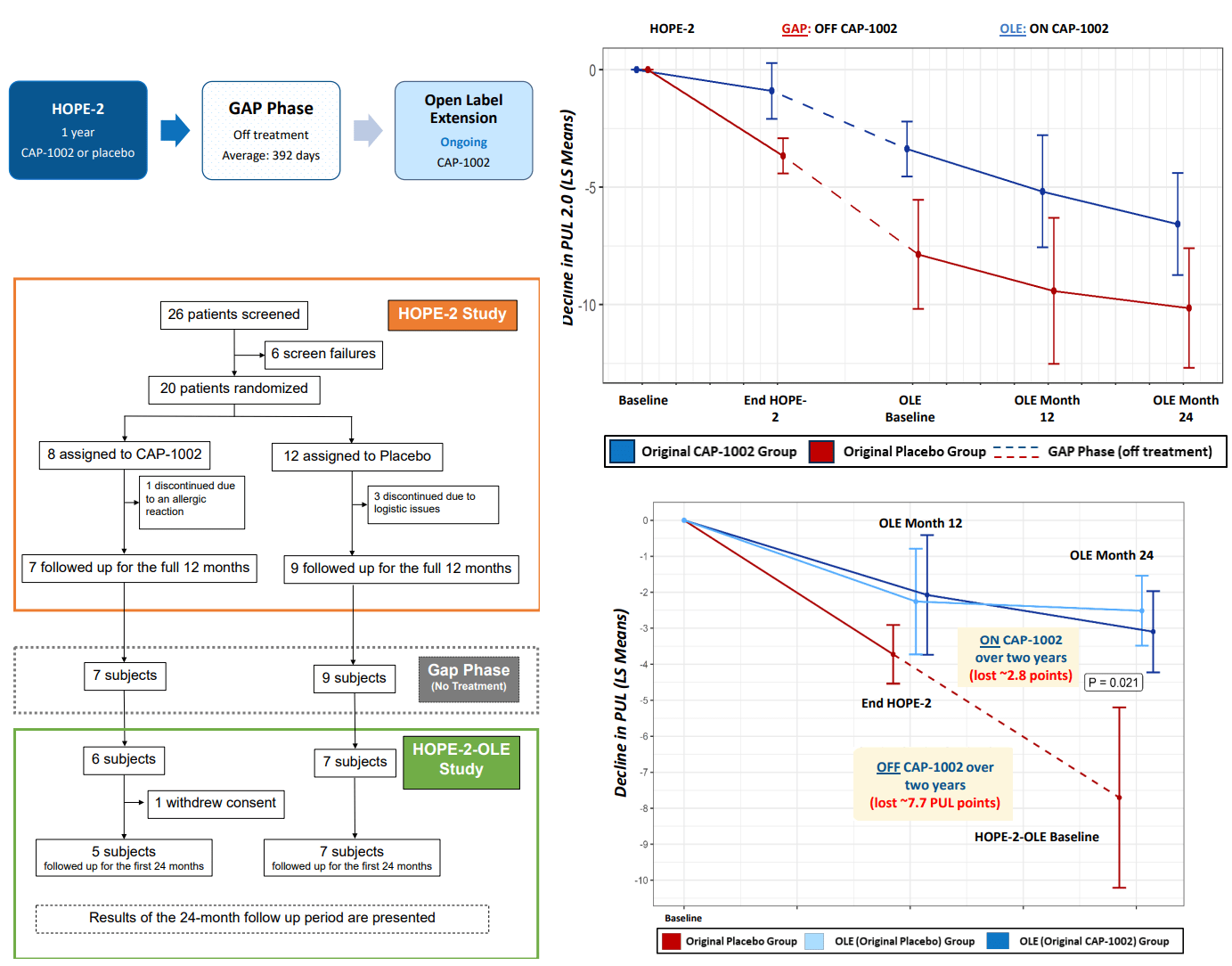
Patients that participated in HOPE-2 were also offered to participate in an open-label extension study for 24 months, after a gap phase (off treatment) of >1 year on average. CAPR recently announced results from the open-label extension of HOPE-2, confirming both durable disease-modifying activity and safety of the treatment. The following are notable from the OLE study;
- Patients on CAP-1002 during HOPE 2 has slower disease progression during the gap phase (suggesting persisting benefit even after withdrawal of treatment).
- Patients regardless of original treatment group in HOPE 2 showed a similar rate of decline; ~2.8 points over 2 years vs. 7.7 point mean decline observed over 24 months in the HOPE-2 placebo patients that went untreated for 24 months during combined 12-month placebo + gap phase (?=4.9 points, p=0.021).
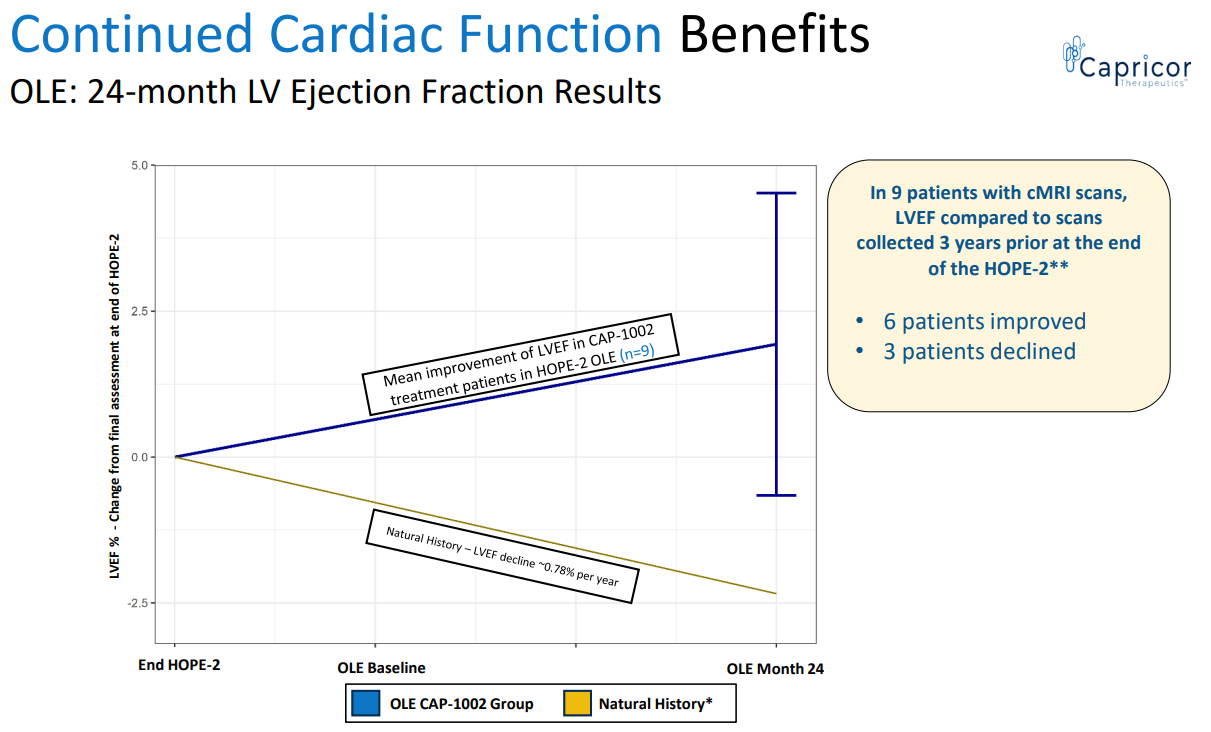
- CAP-1002 resulted in improvement in cardiac function (vs. expected decline).
- CAP-1002 was safe. Re-assuring is also the reportedly lower than expected drop-out rate in ongoing HOPE 3 trial.
Results of the open-label extension of HOPE 2 (CAPR presentations)
{kind=link}
CAP-1002 benefit in cardiac function (CAPR presentation)
{kind=link}
Pathway to approval and commercialization of CAP-1002
Despite hopes for earlier approval by an accelerated approval pathway this does not appear to be the case based on recent update by CAPR, which (combined with a dilutive cash raise) has resulted in >50% dip in CAPR's stock price. This contrasts prior approvals of genetic therapies which were based on increases in dystrophin expression and very small studies. Accelerated approval is based on a surrogate which can be "a laboratory measurement, radiographic image, physical sign or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit". Unfortunately, there is no such established surrogate endpoint for CAP-1002 yet.
The main updates on the regulatory pathway were the following:
- "CAP-1002 product manufactured in San Diego GMP facility not necessary for BLA submission".
- "Ongoing HOPE-3 trial will support BLA submission (n=~58)".
- "Capricor and FDA will continue to discuss other alternative pathways to approval following completion of enrollment in HOPE-3".
Capricor currently maintains two manufacturing facilities; one in Cedars-Sinai Medical Center ("CSMC") and another in San Diego. So far (up to Cohort A of HOPE-3) CAP-1002 was being manufactured in CSMC. However, the CSMC facility "does not meet commercial current Good Manufacturing Practices ("cGMP") standards" based on CAPR's latest 10K. The San Diego facility has been designed to produce commercial-scale GMP CAP-1002 products and to be compliant with U.S., European Medicines Agency, and other international standards.
Based on the update the ongoing HOPE-3 trial now consists of 2 cohorts; Cohort A (which represents the original HOPE-3 trial being conducted with product from the CSMC facility) and a newly announced Cohort B (with product from the San Diego facility). Target enrollment for Cohort A is n=58, reduced from prior target of n=68 "largely due to a very low dropout rate". Completion of enrollment of Cohort A is expected in Q4 2023 and an interim futility analysis is expected in the end of 2023/ beginning of 2024. Considering that the primary efficacy analysis will be conducted at 12 months, topline data should be expected by end of 2024 and CAPR plans to submit a BLA (based on Cohort A) in 2025.
So why the need for Cohort B? Chemistry Manufacturing and Controls ("CMC") issues with cell therapies are complicated. Demonstrating equivalence of the San Diego product to the CSMC product without another clinical trial could prove to be complicated (an issue with cell therapies). Therefore, results of Cohort B appear to be necessary for commercialization by the San Diego facility. Following potential approval based on the CSMC product and completion of Cohort B, CAPR plans to submit a Prior Approval Supplement to shift to commercialization by San Diego facility.
This pathway is much slower than investors had hoped, but according to CAPR registration pathway was shortened by about two years. CAPR still plans to discuss "other alternative pathways" with the FDA, but the practical implications of this statement are unclear.
DMD market opportunity for CAPR
The global DMD market is expected to reach $27.4B - $63.5B by 2030. Capturing even a small fraction of this would mean significant upside for CAPR, which at the time of writing has an enterprise value of about $30M considering recent $23M offering. For comparison, SRPT has at the time of writing an enterprise value of $9.8B (which however is a commercial-stage biotech, has a better pipeline and $1.9 billion in cash, cash equivalents, investments and long-term restricted cash as of June 30, 2023).
In studies from Europe and North America, the incidence of DMD ranges from 1 in 3500 to 1 in 5050 live male births. Considering about 3.7 M and 3.9 M live births per year in US and Europe, respectively, and male-to-female ratio of 1.05 , there should be about 375-630 new cases per year in US and 395-664 new cases per year in Europe (about 770-1300 new cases per year in US and Europe combined). The estimated global prevalence is 7.1 cases (95% confidence interval 5.0-10.1) per 100,000 males. Considering a male population of about 164M in US and 359M in Europe, this corresponds to a DMD prevalence of 11600 in US and 25500 in Europe (CAPR estimates 15000-20000 patients in US and 200000 patients worldwide). Improved life expectancy with new treatments may result in a higher DMD prevalence in the future.
CAP-1002 is currently being developed for patients ?10 years old. Based on above prevalence estimates (and assuming that 30-50% of DMD patients are ?10 years old) I conservatively estimate that 3800-5800 patients would be eligible for CAP-1002. Assuming pricing similar to exon skipping therapies (average $300K/patient/year) this would correspond to sales of $1-1.75B per year. CAPR estimates peak sales of $1B. Considering double-digit royalties this would correspond to peak revenue potential of >100M/year for CAPR based on just US sales. Considering EV/sales ratio of 7.1 (typical for biotechs) this would correspond to an EV of >700M (compared to current EV of $30M).
Additional important considerations for CAP-1002's market potential;
- Above estimate accounts just for US sales.
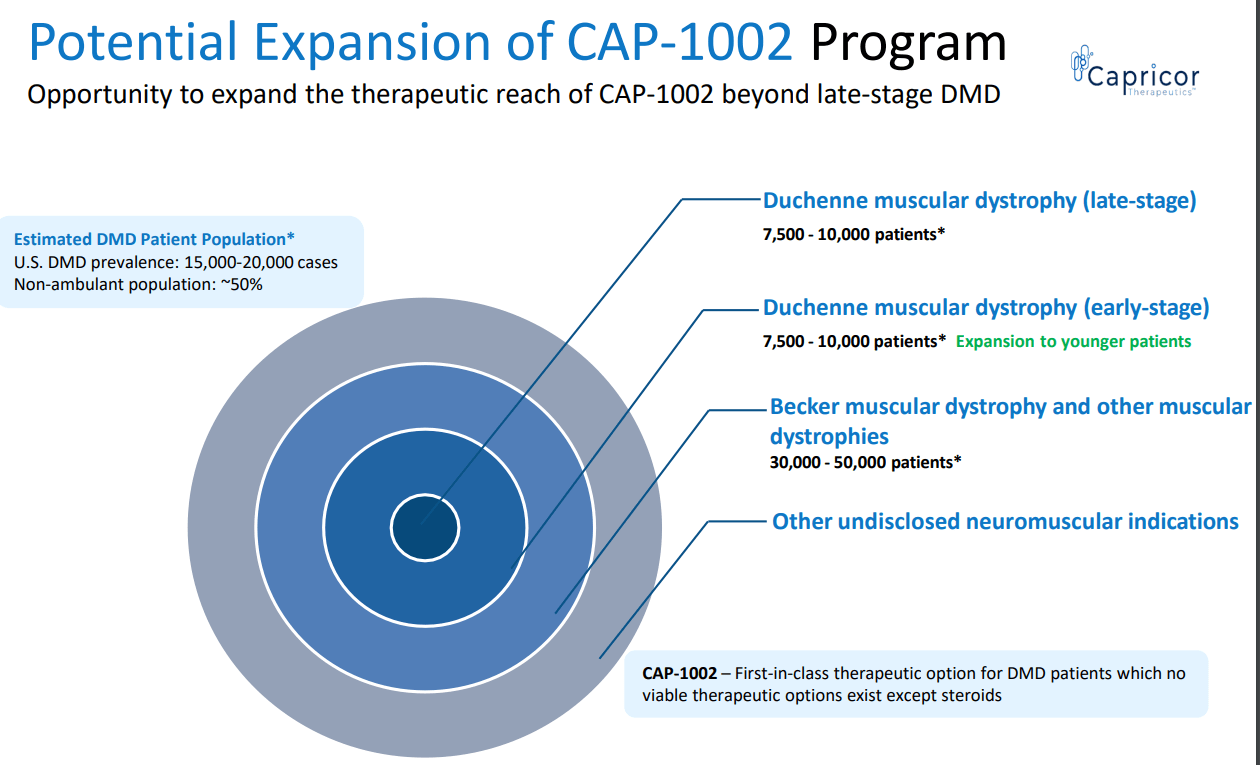
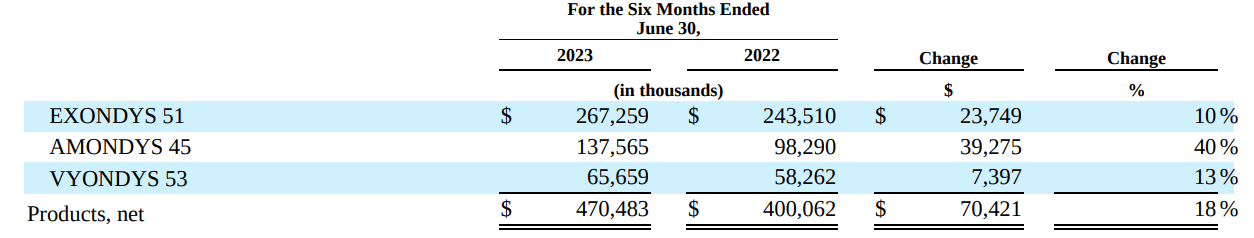
- CAPR's estimation of peak $1B US sales may actually be conservative, especially considering potential expansion (see image below). For comparison, SRPT's net revenue from its exon-skipping therapies (suitable for just 30% of DMD patients) was $470M for the first half of 2023.
- CAPR is currently being developed in older (?10-12 years old) and more advanced (reduced ability to walk or non-ambulatory, >80% were non-ambulatory in HOPE 2) DMD patients. Showing benefit is such patients is a big achievement for CAP-1002 fulfilling an unmet need for these patients. This contrasts clinical trials of approved genetic therapies which enrolled younger ambulant DMD patients. Notably, exon-skipping therapies have no age limitation in their labels (despite enrollment criteria in relevant clinical trials). If CAP-1002 works in patients ?10 years old, I believe that potential expansion to younger patients should be possible and I hope FDA will be lenient in the regulatory pathway for such a label (assuming positive HOPE 3 trial).
- Only 1/3 of DMD patients have mutations amenable to currently available exon-skipping therapies. On the contrary CAP-1002 is not dependent on presence of specific mutations.
- Considering CAP-1002's mechanism of action I agree with CAPR that (provided successful phase 3 and approval) it will be used as a backbone therapy. Therefore, CAP-1002 does not compete with currently approved treatments.
- As highlighted in a recent review article ; "the cell-based CAP-1002 therapy may prove to be a suitable adjunct therapy filling the urgent need for cardiac-specific therapies". Notably, CAP-1002 was originally developed to target DMD cardiomyopathy. In the first HOPE trial CAP-1002 was given by intracoronary administration but showed benefit in both cardiac and skeletal muscle function. Although not the primary outcome of ongoing HOPE-3, CAP-1002's potential to target DMD cardiomyopathy may be a major commercial advantage (cardiomyopathy being the leading cause of death in DMD patients).
- Nippon Shinyaku, exclusive distributor of CAP-1002 in the United States and Japan, has launched Viltepso. Therefore, CAPR will have an experienced partner in the DMD field by the time CAP-1002 is launched.
CAPR's estimations on CAP-1002's target market and expansion potential (CAPR presentation)
{kind=link}
SRPT's net revenue from its exon-skipping therapies (Q2 quarterly filling)
{kind=link}
Nippon Shinyaku partnership
Capricor has entered into two Commercialization and Distribution Agreements with Nippon Shinyaku, appointing Nippon Shinyaku as its exclusive distributor of CAP-1002 in the United States and Japan. Capricor will be responsible for clinical development as well as the manufacturing of CAP-1002. Nippon Shinyaku will be responsible for the distribution of CAP-1002. Capricor will have the right to receive a double-digit share of product revenue and additional development and sales-based milestone payments, if achieved, up to $794 million (up to $705M from US agreement and up to $89M from Japan agreement). Some of these milestones are pre-approval, but the amount of pre-approval milestones is unclear.
Nippon Shinyaku already has an approved DMD exon-skipping therapy (viltolarsen) and is currently developing two more exon-skipping therapies.
DMD therapies in the pipeline
To assess long-term prospects of CAP-1002 it is important to consider DMD treatments currently in clinical development. The following sources were used to identify therapies under development; ClinicalTrials.gov and recent review articles ( 1 , 2 , 3 ). DMD therapies under development include the following;
- AAV microdystrophin gene therapies; Pfizer ( PFE )(in phase 3 stage), Solid Biosciences ( SLDB )(in phase 1/2 ), REGENXBIO ( RGNX )(in phase 1/2 ).
- Exon skipping; REGENXBIO (RGX-253, exon 53, preclinical), Daiichi Sankyo Company ( DSKYF ) (DS-5141, exon 45, phase 2 ), SQY Therapeutics (SQY51, exon 51, phase 1/2 ), Nippon Shinyaku ( NPNKF )(exon 44 phase 2, exon 50 phase 1/2, exon 45/51/55 preclinical), Wave Life Sciences ( WVE )(exon 53 phase 1/2 , other exons in preclinical stage), PepGen ( PEPG ) (exon 51 in phase 2 , exons 44/45/53 in preclinical stage), Cure Rare Disease (targeting various exons, mostly preclinical ), Dyne Therapeutics ( DYN )(exon 51 in phase 1/2 , exons 44/45/53 in preclinical stage), Avidity Biosciences ( RNA )(exon 44 skipping in phase 1/2 ), Entrada Therapeutics ( TRDA )(exon 44/45/50/51 in preclinical/early clinical- stage )
- Therapies targeting inflammation/fibrosis;
- Steroids; vamorolone by ReveraGen Biopharma ( phase 2 ).
- HDAC inhibitor givinostat (oral suspension twice daily); Italfarmaco Group , PDUFA 21-December-2023.
- EDG-5506 ; A small molecule, administered orally once daily, designed to halt the damage in a broad range of rare skeletal muscle disorders. Developed by Edgewise Therapeutics ( EWTX ) currently in phase 2 stage for DMD.
- ATL1102 (antisense inhibitor of CD49d, targeting T cells, anti-inflammatory); Antisense Therapeutics Limited ( phase 2 )
- TAS-205 (HPGDS inhibitor, targets inflammation); Taiho Pharmaceutical ( phase 3 in Japan)
- Pamrevlumab (inhibitor of CTGF, anti-fibrosis); FibroGen ( FGEN ) ( phase 1/2 )
- Cell therapies; CAP-1002 is currently the most extensively studied cell therapy, while many other prior attempts have failed . ENCell is conducting a phase I clinical trial in patients with DMD for its stem cell therapy EN001 based on the transfer of Wharton's jelly (umbilical cord)-derived mesenchymal stem cells.
- In-vivo gene editing technologies (e.g. CRISPR); Delivery of such therapies is typically based on recombinant adeno-associated virus (rAAV) vectors. The use of rAAV vectors to deliver gene therapy is associated with a risk of severe or fatal immune and inflammatory reactions, possibly because high doses of rAAV are needed to deliver sufficient gene therapy to the sizeable amount of muscle tissue in the body. This is illustrated in a recent publication that describes a patient with advanced DMD that had a fatal immune reaction after such a therapy, a risk associated with recombinant adeno-associated virus vectors.
As has been discussed, considering CAP-1002's mechanism of action, genetic therapies (unless curative, which seems unlikely anytime soon) are not likely to be competitive to CAP-1002. In other words CAP-1002 would probably be used as add-on to genetic therapies. CAP-1002 may face competition from other therapies targeting inflammation/fibrosis (most of which are however in earlier stages of clinical development). On the other hand, the several genetic therapies under development pose a long-term risk for SRPT's market share in DMD.
Financials
CAPR's cash, cash equivalents and marketable securities totaled approximately $37.8 million as of June 30 . Total operating expenses for the Q2 2023 were approximately $11.7M (R&D $8.8M, G&A $2.9M). CAPR recently announced a $23 Million Registered Direct Offering (issuance of 4,935,621 shares of common stock, plus warrants to purchase up to 4,935,621 shares at exercise price of $5.70 per share). Based on current operating expenses (about $3.9M per month) cash should be sufficient for a bit more than 15 months, i.e., up to November 2024. Nevertheless, CAPR estimates a cash runaway into 2025, i.e., post Cohort A topline results.
According to CAPR, above guidance does not account for potential milestone payments by Nippon Shinyaku (although the triggers and amount of pre-approval milestone payments are not public). Furthermore, CAPR is looking for a DMD partner in Europe, as well as partnerships for its exosome platform, which could further improve the cash balance of CAPR. Add to that the potential of a priority review voucher after approval (typically worth about $100 million) and CAPR's balance sheet might look much stronger by late 2025. Nevertheless, CAPR will likely have to raise more cash before that. Of note CAPR has a 75 million ATM program (of which 14.6M have been raised up to June 30 2023).
Risks
Risks for CAPR include the following:
- The most important risk is negative/suboptimal results in HOPE-3 (either Cohort A or Cohort B). Notably, HOPE-2 was a very small study (prematurely terminated at n=20 vs. n=84 planned patients due to funding constraints) with some imbalances in baseline characteristics (a bit higher baseline PUL score in placebo). Furthermore, results were marginally better for PUL version 1.2 (original endpoint in HOPE and HOPE 2) compared to PUL version 2.0 (primary endpoint in ongoing HOPE 3). Nevertheless, consistent benefit of CAP-1002 in multiple measures of cardiac function and the open-label extension study, as well as in the preceding HOPE trial suggest a high probability of success of ongoing HOPE 3.
- Delays in the regulatory pathway. Being a cell therapy regulatory hurdles associated with manufacturing are possible. Nevertheless, CAPR has developed a detailed assay for CAP-1002 product identity and potency. CAPR claims that FDA is satisfied with this assay.
- Delays in completion of enrollment of Cohort B of HOPE 3. Notably, in Cohort A first patient was dosed in July 2022 and completion of enrollment is expected in Q4 2024. Therefore, it took about 1.5 years for complete enrollment of Cohort A. Assuming enrollment at a similar pace completion of enrollment should be expected by early/mid-2025 and topline from Cohort B by early/mid-2026.
- Potential competition by new treatments. Considering CAP-1002's mechanism of action the main risk would be from a potentially curative gene therapy (but this is a very speculative long-term risk) as well as other therapies under development targeting inflammation. I believe that for CAP-1002 to be used in combination with therapies like givinostat (PDUFA in December) and EDG-5506 (phase 2 stage) more clinical evidence will be necessary. Therefore, CAPR may face competition from such therapies.
- Need for further dilutive cash raise in the future
Risks for SRPT:
- The main risk for SRPT is failure in one or more of the confirmatory clinical trials. Failure in these trials could result in withdrawal of approved products.
- Another (longer-term) risk for SRPT is emerging competition by other companies developing genetic therapies (discussed in above section). Nevertheless, being first in the market is an important advantage for SRPT.
Conclusions
In my opinion, the dip in CAPR stock price following updates on regulatory pathway and the offering represents a buying opportunity. Based on promising Phase 2 results (published in Lancet) there is high probability of success for ongoing phase 3 trial. Furthermore, CAP-1002 does not compete with existing therapies and its adoption by healthcare professionals and patients would likely be quick (assuming positive phase 3 and approval). As far as SRPT is concerned it is a leader in the field with several approved products, increasing revenues, promising pipeline and great cash balance. Major risks for SRPT include failure in confirmatory trials and competition by many other companies developing genetic therapies for DMD. Note that investing in CAPR (a clinical-stage micro-cap) is a high-risk investment.
Your feedback is appreciated
Please comment below if you have any feedback (positive or negative), if you spot any mistakes, or if you believe I missed something important in my analysis.
For further details see:
Capricor Therapeutics, Sarepta Therapeutics: A Promising Clinical-Stage Biotech And A Commercial-Stage Leader In Duchenne Muscular Dystrophy